Calling HLA-A and B Alleles
PharmCAT supports HLA allele data with the following option, which is discussed in more detail below:
- using a different tool to determine HLA alleles, which can be imported into PharmCAT to provide a translation of the phenotypes and recommendations.
Problems with calling HLA from VCF
The HLA region is highly polymorphic and complex region filled with genes, retrotransposons, transposons, regulatory elements, and pseudogenes that make mapping using with short sequence reads problematic. The current human reference genomes are linear references (hg19 and hg38); therefore, a single allele is designated as a reference allele to serve as part of the human genome causing mapping difficulties when the allele deviates from the reference too much. More recent efforts such as the latest hg38 have made great efforts to improve mapping by including several alternative contigs for HLA to the reference. Calling HLA from a VCF is difficult given that most of the variation necessary to type an HLA allele can be missing from the files, and methods to call HLA from a VCF file generally rely on using well known haplotype tagging SNPs in the population or HLA imputation using population references. We recommend that you use targeted high resolution typing for HLA or calling from alignment files/raw sequence reads containing both mapped and unmapped reads.
HLA Imputation
We tested imputation accuracy using 258 in house samples for which we had targeted HLA sequencing and low-pass whole genomes (~2.54X). To impute, we used the Michigan Imputation Server Four-digit Multi-ethnic HLA v1 (2021) panel, which uses the SNP2HLA program as part of the HLA-TAPAS software to call HLA. The reference panel used contains 36,586 HLA haplotypes belonging to a multi-ethnic cohort. For our testing purposes, we looked at 4 important PGx HLA alleles (HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, HLA-B*58:01). All 4 alleles passed QC as described in Luo et al. 2021. A summary of our results are as follows:
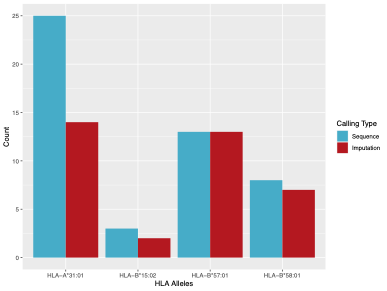
| HLA Allele | Sequence | Imputed |
|---|---|---|
| HLA-A*31:01 | 25 | 14 |
| HLA-B*15:02 | 3 | 2 |
| HLA-B*57:01 | 13 | 13 |
| HLA-B*58:01 | 8 | 7 |
Our results indicate that HLA B allele imputation was more accurate than the single HLA-A allele tested. While imputation is possible from a VCF, we do NOT recommend imputing HLA from a VCF as there are many factors that contribute to inaccuracy (e.g., marker density, an appropriate reference panel, etc.). Although the methods and reference panels have drastically improved over the past few years, we still have reservations based on our own internal testing of the accuracy of allele typing. Instead, we recommend that you call HLA directly from either whole exome or whole genome data.
Typing HLA from Whole Genome or Whole Exome Datasets.
For demonstration purposes, we will use the commonly used program Optitype, available through the Nextflow analysis pipeline HLA Typing. To install Nextflow, we recommend you follow the conda installation for Nextflow found on the website. Nextflow facilitates the use of reproducible pipelines while also enabling the use of various software packages in various computational environments. Optitype is a freely available software package used to call HLA Class I alleles. Please note that the references are based on the IMGT/HLA Release 3.14.0, which does not affect the HLA alleles we tested for PGx. Nextflow is currently working on building a solution to bring in newer IMGT releases. If you are looking to call HLA Class II alleles, please look at HLA-LA but note that they only type HLA based on the protein binding domains and therefore are only able to output G-group resolution.
To type using Optitype for HLA calling from Whole Genome Data or Whole Exome Data, you have two options to work from.
-
You can work directly with the RAW FASTQ files output from the sequencer.
-
You can work directly with the BAM/CRAM files output after alignment.
Typing from FASTQ will only work if you only have two FASTQ files to work from. In certain cases where more than the pair are generated such as from running on different lanes, we recommend you align them and work from the alignment files, ask the sequencing center to provide files with no lane splitting, or attempt to concatenate them yourself using bash commands such as cat. We are not affiliated with either Nextflow or Optitype and we suggest you contact them for questions or for troubleshooting.
HLA Typing using Optitype from raw FASTQ Files
For our test, we used the 1000 genome HLA calls that are available through The International Genome Sample Resource and described in Abi-Rached et al, 2018. These HLA calls were generated using the commercial software PolyPheMe from WGS. We picked several samples across different populations that had one of our alleles of interest: HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, HLA-B*58:01.
In total, we tested 203 samples that intersected with those in Abi-Rached et al, 2018 and had exome sequence FASTQ available. These exome sequences were generated for phase 3 of the 1000 genomes and averaged 65.7x depth.
For demonstration purposes, we will use HG00122. To download the files to your computer or cluster, you can use a command such as wget or browse the ftp server using Fetch or Cyberduck.
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR711/SRR711355/SRR711355_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR711/SRR711355/SRR711355_2.fastq.gz
These commands will download SRR711355_1.fastq.gz and SRR711355_2.fastq.gz compressed FASTQ files. It is necessary to download both to have both the forward and reverse reads.
You can follow the Full Sample sheet documentation and the usage docs to see how to proceed to generate a sample list and submit for Nextflow. Generally, the sample sheet will consist of a CSV file with the following fields:
| sample | fastq_1 | fastq_2 | seq_type |
|---|---|---|---|
| HG00122 | /path/to/file/HG00122/SRR711355_1.fastq.gz | /path/to/file/HG00122/SRR711355_2.fastq.gz | dna |
These sample sheets can consist of a single sample or multiple samples. Regardless, each sample will be processed separately and in its own individual folder. You can save your document as: sample_sheet.csv
If you followed the documentation and installed nextflow using conda, you should activate your environment.
conda activate env_nf
You can submit your file either as a script or through an interactive session:
nextflow run nf-core/hlatyping --input /path/to/file/sample_sheet.csv --outdir results -profile singularity
You can read the usage docs to see what parameters you have available in the Nextflow pipeline. In this case, I am running their hlatyping pipeline, inputting the path to the sample sheet, specifying the output directory, and using singularity based containers. There are other options available to suit your needs such as Docker and Conda.
Running the command above will a folder in the output results with your sample name. The folder includes two documents: 1) a pdf of the coverage across the Class I HLA Alleles, 2) the results of the typing in a tab delimited file.
Note, of the 203 samples tested, Optitype misassigned an HLA to 3 of the samples tested, e.g., HG00140, due to sequencing coverage issues. It is worth noting that these calls had a much lower amount of sequencing reads in the HLA region (550-650) than the average amount of the cohort (1068). Also, based on the plots, there were gaps in the HLA genes being typed, which could have affected the calling.
| HLA Allele | OPTITYPE CALLS | 1KG OFFICIAL CALLS |
|---|---|---|
| HLA-A*31:01 | 47 | 48 |
| HLA-B*15:02 | 39 | 39 |
| HLA-B*57:01 | 54 | 54 |
| HLA-B*58:01 | 67 | 69 |
Regardless of whether it is a single sample per sheet or multi-sample per sample sheet, Optitype will generate individual files for each sample. Nextflow It will generate a plot showing the overall coverage of the allele and a TSV (tab-separated values) file with the HLA allele calls. It is always a good idea to check and ensure you had good coverage across the HLA region.
HLA from WES and WGS Alignment Files
If you have BAM or CRAM files available to you and not the original FASTQ files, your workflow will look something like this. Working backwards from the BAM/CRAM files requires a lot more computational power, time, and disk space. Most likely, you will need to use a cluster to run the following.
For testing purposes, we accessed the 30X WGS CRAM (Compressed Reference-oriented Alignment Map) files through the Globus Endpoint. Since Optitype takes either FASTQ files (Raw Sequence files with Quality Scores) or BAM files (Binary Alignment Map) as input, we converted the CRAM files to BAM files using Samtools. CRAM files are a lot smaller than BAM files, but do not contain all the information in a BAM file and therefore requires the reference FASTA file used in the alignment process. Converting a CRAM to BAM is easy, but be prepared for a much larger disk space footprint of the BAM file:
samtools view -b -T <refgenome.fa> -o <output_file.bam> <input_file.cram>
For a 1000 genome use case scenario, such as HG00140.final.cram, we can convert it as:
samtools view -b -T GRCh38_full_analysis_set_plus_decoy_hla.fa -o HG02420.final.bam HG02420.final.cram
You can visit the samtools documentation if you would like to modify the commands or look into directly converting the CRAM file to FASTQ files directly. Once converted to BAM files, you can follow the Full Sample sheet documentation and the usage docs to see how to proceed to generate a sample list and submit for Nextflow.
| sample | fastq_1 | fastq_2 | bam | seq_type |
|---|---|---|---|---|
| HG02420 | /path/to/file/HG02420.final.bam | dna |
The first steps that Nextflow does is convert the BAM files back into 2 FASTQ files to realign the reads with Optitypes references. Using the high-coverage data for a minimum of 10 individuals known to carry a PGx HLA allele, our results indicated a 1:1 relationship for the 4 PGx HLA alleles tested. Similarly, HLA-LA also had a 1:1 relationship for those specific alleles once we truncated the G-group resolution to a 2-field, but the G-group nomenclature also includes other HLA proteins that share the same nucleotide sequence for the protein binding domain (exons 2 and 3 for HLA class I and exon 2 only for HLA class II alleles).
For example: A*33:01:01G includes alleles such as: A*33:01:01:01, A*33:01:01:02, A*33:01:01:03 which share the same protein, but it also contains A*33:220, A*33:222, A*33:228, A*33:234, which have different proteins, but share the same nucleotide sequence in exons 2 and 3 as the others.
Although we did not simulate coverage, a recent study by Thuesen et al. 2022, evaluated software performances of the most common HLA calling software, including Optiype, at different simulated coverage and degraded conditions such as those with aDNA. While performance was relatively stable across different coverages for Whole Exome Sequence Data and Optitype, we still recommend working with as high coverage as possible.
Working with HLA in PharmCAT
PharmCAT supports incorporating results from your favorite HLA programs for phenotype translations through “outside calls.”
To incorporate the outside calls, you would run PharmCAT as you normally would, and add the -po flag which signals the program to look for an external call file. Please note that PharmCAT requires that your HLA call have only two fields for phenotype translations. Therefore, if your external calls have more than two fields, you should truncate your output to only two fields. For more information on HLA nomenclature and what the each field means in HLA, please visit the page for Nomenclature for Factors of the HLA System page. For example:
# run the HLA calls with a VCF
java -jar pharmcat.jar -vcf test.vcf -po /path/to/test_sample_hla.txt
# only generate a report for the HLA calls
java -jar pharmcat.jar -phenotyper -reporter -po /path/to/test_sample_hla.txt
test_sample_hla.txt should be a tab-delimited file where the first column is the gene name and the second column the HLA diplotype:
HLA-A *32:01/*68:03
HLA-B *07:02/*35:01
For more information:
-
see Running PharmCAT for details on the -po flag
-
see Outside Call Format for details on the outside call file
Formatting Optitype output for PharmCAT
We recommend calling HLA from Whole Genome Sequencing or Whole Exome Sequencing (WGS or WES) from BAM or FASTQ files using software similar to Optitype. We also recommend that you use Optitype through the Nextflow pipeline for reproducibility and ease of use and installation. This is especially ideal if you are already using the pipeline to call CYP2D6 using StellarPGx.
While this tutorial is specific to Optitype, it can be modified for integrating HLA calls from other programs. When you run Optitype, whether a single sample or multiple sample run, the program outputs a single file TSV file per individual, which looks like:
A1 A2 B1 B2 C1 C2 Reads Objective
0 A*32:01 A*68:03 B*07:02 B*35:01 C*07:02 C*07:02 10191.0 9915.832999999959
Each column represents a separate HLA allele call. To convert this into a format digestible by PharmCAT, you must reorder the TSV file. This can be done using a simple Python script. The following is a simple script to parse Optitype's output and output a file format usable for PharmCAT. Currently, we only support HLA A and B calls in PharmCAT, so therefore only those two genes are output.
#!/usr/bin/env python
import pandas as pd
import sys
import os
# Prompt for the Sample Name, which will be used as the file name and input file path to the TSV file
sample_name = sys.argv[1]
input_path = sys.argv[2]
# Load TSV file as a dataframe
df = pd.read_csv(input_path, sep="\t")
# Reformat Data
a_values = df.iloc[0, 1:3].str.replace("[AB](?=\*)", "", regex=True).str.cat(sep="/")
b_values = df.iloc[0, 3:5].str.replace("[AB](?=\*)", "", regex=True).str.cat(sep="/")
# Create new dataframe with reformatted data
new_df = pd.DataFrame([["HLA-A", a_values], ["HLA-B", b_values]])
# Export the data frame into a Tab-delimited file
new_df.to_csv(f"{sample_name}_HLA.txt", sep="\t", index=False, header=False)
Below is a usage example of how it would be called from through the command path:
python3 Optitype_to_PharmCAT.py <sample_ID> <input file path or input file>
python3 Optitype_to_PharmCAT.py test_sample test_sample_results.tsv
The output file test_sample_hla.txt should look like:
HLA-A *32:01/*68:03
HLA-B *07:02/*35:01
This script can be used in conjunction with bash scripting to automate the processing of multiple sample files, where you can use the sample names as variables.
We are not responsible or can guarantee how these programs will perform and should primarily be used for research purposes. We suggest you consult a clinical laboratory if you need clinical-grade HLA typing, particularly high-resolution typing based on NGS. Generally, clinical laboratories can provide an HML file, and we will explore how to bring that data into PharmCAT in the future.